About me
My name is Khoa, and I am a senior, double-major in Quantitative Economics and Data Science. My senior capstone project is about computational drug development, focusing on the application of deep learning in predicting the solubility of drug compounds from the public ZINC online database.
My project
Abstract
Drug discovery and development is a costly and time-consuming process, taking up to billions of dollars and 12-15 years from basic research to FDA approval. Early stage discovery involves intensive search through an enormous database of molecules and analysis of their quantitative structure-activity relationships to determine their physicochemical properties. Important features like absorption, distribution, metabolism, and excretion (ADME) are extracted to measure how these compounds interact with the human bodies. At its root, this is an optimization problem in which researchers try to identify the “best” compounds with desired properties to be qualified for clinical development to produce a safe and cost-effective drug. Nowadays, with stronger computation power, the process can be sped up significantly with artificial intelligence. Many deep learning models have demonstrated highly accurate predictions on the ADME properties of drug-like small molecules. In particular, graph neural networks (GNN) are shown to learn effectively graph-based molecular representation. This paper examines the feasibility of several state-of-the-art graph neural networks on predicting the solubility of commercially available compounds in the ZINC database. The experiment indicated that each model’s performance was significantly improved through training. The results suggested promising applications of deep learning in reducing the time and cost of the drug development process in the foreseeable future.
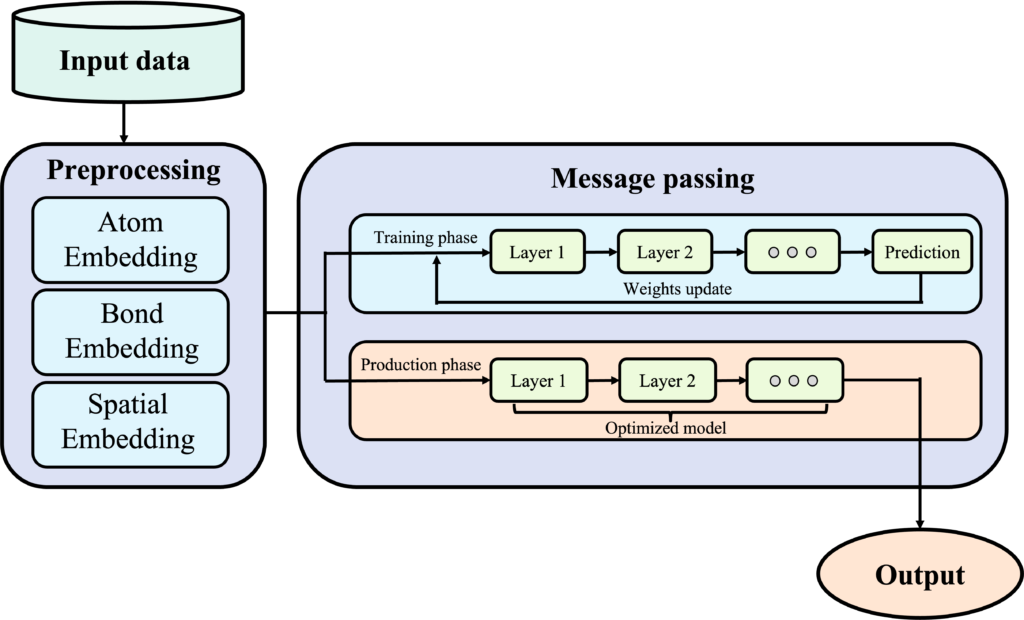
Data diagram
Gitlab project
Paper
Software demonstration video
Poster
